User's Guide: METASOFT
Important: User interface has been changed since v2.0.
Usage:
usage: java -jar Metasoft.jar [options]
-input <FILE> Input file (Required)
-output <FILE> Output file (default='out')
-log <FILE> Log file (default='log')
-pvalue_table <FILE> Pvalue table file (default='HanEskinPvalueTable.txt')
-lambda_mean <FLOAT> (Random Effects) User-specified lambda for mean effect part
(default=1.0)
-lambda_hetero <FLOAT> (Random Effects) User-specified lambda for heterogeneity
part (default=1.0)
-binary_effects Compute binary effects model p-value (default=false)
-binary_effects_sample <INT> (Binary effects) Number of importance sampling samples
(default=1,000)
-binary_effects_p_thres <FLOAT> (Binary effects) P-value threshold determining if we will
use large number of samples (default=1E-4)
-binary_effects_large <INT> (Binary effects) Large number of importance sampling samples
for p-values above threshold (default=100,000)
-mvalue Compute m-value (default=false)
-mvalue_method <METHOD_NAME> Which method to use to calculate m-value between 'exact' and
'mcmc' (default=exact)
-mvalue_p_thres <FLOAT> Compute m-values only for SNPs whose FE or RE2 p-value is
below this threshold (default=1E-7)
-mvalue_prior_beta <ALPHA BETA> Alpha and Beta value for Beta dist prior
Betadist(alpha,beta) for existence of effect
(default=1.0,1.0)
-mvalue_prior_sigma <FLOAT> Sigma value for normal prior N(0, sigma^2) for effect
(default=0.2)
-mcmc_sample <INT> (MCMC) Number of samples (default=10,000)
-mcmc_burnin <INT> (MCMC) Number of burn-in (default=1,000)
-mcmc_max_num_flip <INT or FLOAT> (MCMC) Usual move is flipping N bits where N ~
U(1,max_num_flip). If an integer value i >= 1 is given,
max_num_flip = i. If a float value 0 < k < 1 is given,
max_num_flip = k * #studies. (default=0.1)
-mcmc_prob_random_move <FLOAT> (MCMC) Probability that a complete randomization move is
suggested (default=0.01)
-seed <INT> Random number generator seed (default=0)
-verbose Print RSID verbosely per every 1,000 SNPs (default=false)
-help Print help
BE is another kind of random effects model assuming that the effects either exists or not in the studies. Our simulations show that BE is often more powerful than RE2. However, BE requires importance sampling to calculate p-value and is slower than RE2. To use BE, use option -binary_effects. We implemented an adaptive sampling that performs BE with smaller number of samples first (-binary_effects_sample option), and if the calculated p-value exceeds a threshold (-binary_effects_p_thres option), then a larger number of samples are used for higher accuracy (-binary_effects_large option).
To compute m-values, use option -mvalue.
M-values require two priors: Beta distribution prior for the presence/absence of effect
(specified by -mvalue_prior_beta option),
and normal distribution prior for the effect size
(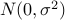 where
where 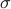 is specified by -mvalue_prior_sigma option).
The default for Beta distribution prior is uniform distribution (
is specified by -mvalue_prior_sigma option).
The default for Beta distribution prior is uniform distribution (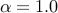 and
and 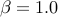 )
and the default for is 0.2.
Since we assume a normal prior for effect size, for quantitative traits, the value of
has to be chosen carefully considering the unit of the data.
One can choose a value based on the data in an empirical Bayes style,
or a rough and ad-hoc approach is just using the default value (0.2) designed for dichotomous traits.
)
and the default for is 0.2.
Since we assume a normal prior for effect size, for quantitative traits, the value of
has to be chosen carefully considering the unit of the data.
One can choose a value based on the data in an empirical Bayes style,
or a rough and ad-hoc approach is just using the default value (0.2) designed for dichotomous traits.
The computation time is exponential to the number of studies. If the number of studies is small (e.g. <10), an exact calculation is feasible (-mvalue_method exact option). If the number of studies is large, MCMC should be used (-mvalue_method mcmc option). The user can specify the number of burnin (-mcmc_burnin) and the number of samples (-mcmc_sample). The MCMC move is flipping the existence of effect for N studies where N is sampled from Uniform(1, max_num_flip) where max_num_flip is specified with -mcmc_max_num_flip option.
File Format
Input File format:
Rows are SNPs.
1st column is RSID.
2nd and 3rd columns are effect size (beta) and its standard error of study 1.
4th and 5th columns are effect size (beta) and its standard error of study 2.
6th and 7th columns are effect size (beta) and its standard error of study 3.
and so on…
Please write “NA” for missing beta and its standard error.
rsAAAAAA study1beta study1stderr study2beta study2stderr study3beta study3stderr rsBBBBBB study1beta study1stderr study2beta study2stderr study3beta study3stderr rsCCCCCC study1beta study1stderr study2beta study2stderr study3beta study3stderr
Example Running Command:
java -jar Metasoft.jar -input example.txt
Output File:
The columns in output file are as follows
| Column Num. | Column name | Description |
| 1 | RSID | SNP rsID |
| 2 | NUM_STUDY | Number of studies used in meta-analysis for the SNP |
| 3 | PVALUE_FE | FE P-value |
| 4 | BETA_FE | Estimated Beta under FE |
| 5 | STD_FE | Standard error of BETA_FE |
| 6 | PVALUE_RE | RE P-value |
| 7 | BETA_RE | Estimated Beta under RE |
| 8 | STD_RE | Standard error of BETA_RE |
| 9 | PVALUE_RE2 | RE2 P-value |
| 10 | STAT1_RE2 | RE2 statistic mean effect part |
| 11 | STAT2_RE2 | RE2 statistic heterogeneity part |
| 12 | PVALUE_BE | BE P-value (“NA” if -binary_effects option is not used) |
| 13 | I_SQUARE | I-square heterogeneity statistic |
| 14 | Q | Cochran's Q statistic |
| 15 | PVALUE_Q | Cochran's Q statistic's p-value |
| 16 | TAU_SQUARE | Tau-square heterogeneity estimator of DerSimonian-Laird |
| 17 … 17+NUM_STUDY-1 | PVALUES_OF_STUDIES | P-values of each single studies |
| 17+NUM_STUDY … 17+2*NUM_STUDY-1 | MVALUES_OF_STUDIES | M-values of each single studies (“NA” if -mvalue option is not used) |
Log File:
The data reported in log file are as followsNumber of SNPs analyzed
Number of studies
Calculated genomic-control inflation factor for mean effect part of RE2 statistic
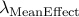
Calculated genomic-control inflation factor for heterogeneity part of RE2 statistic
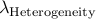
We provide a simple method for correcting for population structure for RE2.
First, put all SNPs into a single file.
Then apply our software without using -lambda_mean or -lambda_hetero options.
Look at log file to see what are the inflation factors for both the mean effect
part and heterogeneity part of RE2 statistic,
which we denote and .
Then, apply our software one more time using
the options -lambda_mean and -lambda_hetero .
Often, the user wants to apply genomic control not only to the meta-analysis p-values but also to each single study before the user uses them in the meta-analysis. This can be easily done by, in the input file, increasing the standard errors by the square root of the calculated inflation factor for each study.
Format converter
Plink format converter usage:
python plink2metasoft.py outputfile plink_assoc_file1 plink_assoc_file2 plink_assoc_file3 ...
Our software converts multiple .assoc files from plink to the input format of METASOFT. If .assoc files include both A1 and A2 columns, our converter can detect simple strand flips. There are three output files: .meta file is the formatted file, .mmap file includes SNP map information, and .log file includes log information such as strand flipped SNPs, excluded SNPs, and reasons for exclusions. Python and R must be installed in the system to use our converter.