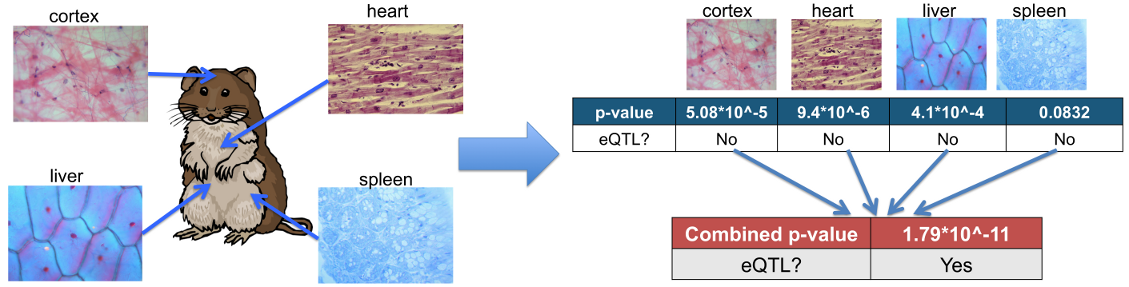
Instruction step 1: prepare multiple tissue dataset
Users need to provide a few files before using Meta-Tissue. Those files can be categorized into 3 groups: 1) Information on which tissues are collected from each sample (called "tissue information"), 2) Gene expression data, 3) Genotype data. Meta-Tissue expects the following files for each category (all of following example files are stored in "example/1_orig_input/" folder in the software package).
1. Tissue information file: this file contains information on which tissues are collected from each sample. Here is one such file (tissue_info.txt)
|
1. First line always starts with "#TISSUE" followed by tissue names (e.g. "cortex," "heart," "liver," and "spleen")2. From the second line, information on which tissues are collected from each sample is specified2.1 First column is sample ID (e.g. "a" "b" "c" ...)2.2. Second and later column is 1 if the tissue was collected from this individual or 0 otherwise. For example, cortex, heart, and liver were collected from sample "a" while liver and spleen were collected from sample "b"3.IMPORTANT!!! Columns must be separated by a whitespace (space or tab), but multiple whitespaces are NOT allowed and will cause errors. |
2. Gene expression data: there are 3 different types of files specifying information on gene expression data.
2.1 Gene expression file: This specifies gene expression level for each sample measured on multiple probes. Meta-Tissue expects one gene expression file for each tissue. So, if there are 4 tissues collected, then there must be 4 files. The format of each gene expression file is as follows (cortex.txt):
|
1. First line lists sample IDs collected for this tissue.2. Please note that samples that are not collected for this tissue must not be listed in gene expression file. For example, sample "b" is not listed in this file (cortex.txt) since cortex tissue was not collected from sample "b" (see above tissue information file).3. IMPORTANT!!! The order of sample IDs must match the order of sample IDs in the tissue information file. For example, "a d e c" in the first line of gene expression file is incorrect because "c" comes before "d" in the tissue information file.4. Second line and later specify gene expression level for each sample. Each row is a probe for gene expression and each column corresponds to a sample. |
2.2 Gene expression list file: This specifies where the above gene expression files are stored. Specifically, it lists the full path to the above files. Here is one example (gene_list.txt)
|
1. Each line lists the full path to the gene expression file.2. IMPORTANT!!! The order of gene expression files in this file must be the same as the order of tissues listed in the first line of tissue information file (tissue_info.txt). In the above example, the order of tissues was "cortex heart liver spleen," and the order of expression files listed here is also "cortex heart liver spleen."3. IMPORTANT!!! Always use the full or absolute path (e.g. "/usr/home/[user_id]/") when you specify files in Meta-Tissue. Relative path (e.g. "../") or tilde ("~") may cause errors. |
2.3 Probe information file: This specifies information on each probe (probe_info.txt)
|
1. Each line lists information on each probe.2. The first column must be probe ID.3. The second column must be "chr[chr_number]" where chr_number can be 1-22, X, Y, and XY.4. The third column must be position of probes (starting position). |
3. Genotype data: Meta-Tissue currently supports EIGENSTRAT format only. Other formats (PLINK and VCF) will be supported in future release. To prepare genotype data in EIGENSTRAT format, please use "convertf" tool in EIGENSTRAT software. Here is a command file that converts PLINK file to EIGENSTRAT format.
|
1. Use command "convertf -p (command_file)"2. You can also use PLINK binary format. Specify [PLINK].bed in genotypename, [PLINK.bim] in snpname, and [PLINK.fam] in indivname fields.3. IMPORTANT!!! The order of samples in PED file must match the order of samples in tissue information file.4. Sample output files (geno.eigenstrat, ind.txt, and snp.txt) are stored in the example folder. |
4. Covariate data: Since v0.4, Meta-Tissue supports covariates, which are included as fixed effects in the linear mixed models.
|
1. The format of covariate file is that each line is each individual and each column is each covariate.2. The first column is ID of each individual, and the second column is the first covariate, the third column is the second covariate, and so on.3. IMPORTANT!!! The order of individuals must match the order of individuals in tissue information file.4. No missing values are allowed in the covariate file. |