Instruction step 3: Run MetaTissueMM (Mixed Model) to obtain estimates of effects
The third step in Meta-Tissue is to run software called MetaTissueMM to compute estimates of effects and their standard errors. These estimates are required for meta-analysis we will perform in step 4. MetaTissueMM computes the estimates by taking into account the fact that multiple tissues are collected from the same individual.
MetaTissueMM is a linux program and cannot be executed in Windows or Mac. Here is its usage. Please note that since v0.3, all options are changed to long option
|
Here is more information on above parameters
|
When running MetaTissueMM on a large GWAS dataset (more than a thousand samples in all tissues with more than 500K SNPs), it is recommended that you parallelize the analysis using the cluster of nodes. Users can achieve this by running MetaTissueMM on a subset of SNPs (but for all gene expression probes), and "--start_snp_index" and "--end_snp_index" options specify which SNPs to analyze. The index starts with 0 (zero), and "--start_snp_index" option is inclusive while "--end_snp_index" option is exclusive. This means that if users want to run MetaTissueMM on the first 1,000 SNPs, they need to specify "--start_snp_index 0 --end_snp_index 1000" and for the next 1,000 SNPs, they specify "--start_snp_index 1000 --end_snp_index 2000" and so on. |
|
MetaTissueMM generates 6 files as output.
This is input file to Metasoft. It contains estimates of effects and their standard errors for pairs of SNP and gene expression. Each line specifies each such pair in all tissues.
This is a shell script that runs Metasoft automatically with the above input file. This will be discussed more in Step 4.
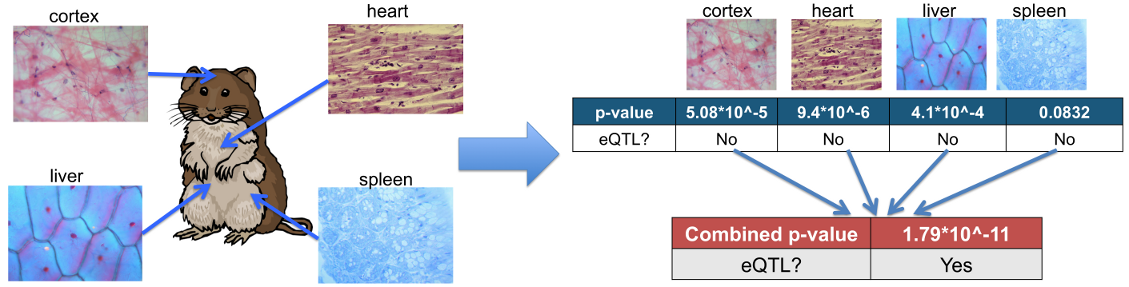
This is output of "Tissue-By-Tissue" approach that computes a p-value for each tissue separately. Each line is each pair of SNP and gene expression and there are (T+1) columns where there are p-values for T tissues (+ ID in the first column). The order of p-values is the same as the order of tissues in tissue information file. Users should use this result to detect tissue-specific eQTLs (for more info, refer to our manuscript)
This is a log file of MetaTissueMM.
This specifies correlation among beta.
This specifies variance explained by the random effects. Only used when --heuristic is enabled |
|
These options are related to Metasoft. [Metasoft dir] is the "Metasoft" folder in software package. Please specify the full path to this folder! When --no_mvalue is specified, Metasoft does not compute m-value for each tissue. |
|
Turn on dosage mode. Genotype file contains dosage information created with -x option using MetaTissueInputGenerator. |
Here is a sample command using the input files in "example/2_MetaTissue_input/" folder and using "example/3_MetaTissue_output/" as output folder in the software package.
|